
PetaChem
¿Qué es PetaChem?
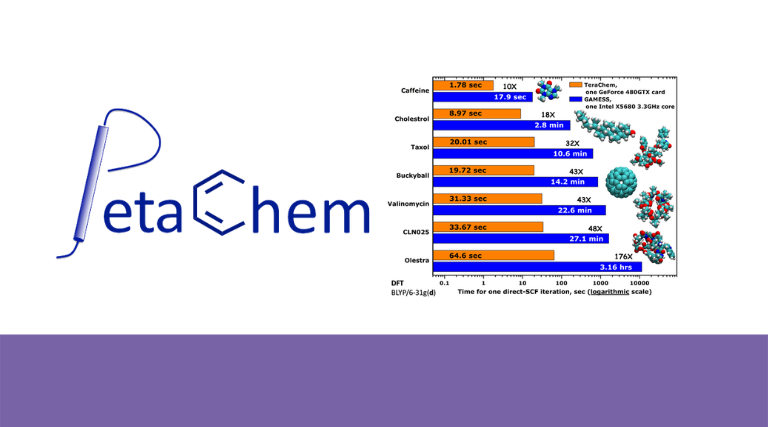
PETA CHEMPetaChem, LLC está dedicado a permitir que la química cuántica y la dinámica de primeros principios para materiales moleculares y moléculas biológicas. Nuestro enfoque es la velocidad y lo logramos a través rediseño de algoritmos modernos para procesadores de flujo, como las arquitecturas GPU CUDA de NVIDIA.
¿Qué puede ayudarte PetaChem?
Es un software de química cuántica de propósito general diseñado para ejecutarse en arquitecturas de GPU NVIDIA en un sistema operativo Linux de 64 bits. Algunas de las características de TeraChem incluyen:
Soporte completo para las GPU NVIDIA GeForce / Tesla (Fermi, Kepler, Maxwell y Pascal)
Restricciones, restricciones y restricciones de concha abierta Hartree-Fock y cálculos de energía y gradiente de Kohn-Sham basados en cuadrícula
Soporte completo de funciones básicas tipo s, p y d
Diversas funciones DFT, incluidas las funciones corregidas por rango y atenuadas de Coulomb (BLYP, B3LYP, PBE, PBE0, ωPBE, ωPBEh, ωB97, ωB97x, camB3LYP, etc.) y cuadrículas DFT (800 – 80,000 puntos de rejilla por átomo)
Rejillas DFT estáticas y dinámicas.
Corrección de dispersión empírica (DFT-D3 y DFT-D2)
Teoría funcional de la densidad dependiente del tiempo (TDDFT) y CI Singles (CIS)
Optimización de la geometría (L-BFGS, gradiente conjugado, descenso más pronunciado) y búsqueda del estado de transición
La optimización puede llevarse a cabo en coordenadas cartesianas o internas como se especifica en el archivo de inicio (todas las geometrías de entrada se proporcionan en cartesianos). La transformación de coordenadas cartesianas → internas → cartesianas se realiza automáticamente siempre que sea necesario.
Optimización restringida con átomos congelados, longitudes de enlace restringidas, ángulos y diédricos.
Dinámica molecular ab initio (conjuntos NVE, NVT)
Dinámica reversible del tiempo de Born-Oppenheimer
Condiciones de contorno esféricas
Soporte de sistemas de múltiples GPU
Precisión de precisión simple / dinámica / doble
Tratamiento QM / MM de moléculas de agua circundantes usando el campo de fuerza TIP3P
Análisis orbital de enlace natural a través de la integración con NBO6
Polarizabilidades para los métodos HF y DFT de capa cerrada

